Matthew Chambers

E-mail: chambers@lsu.edu
B.A.: Cornell University
Ph.D.: Massachusetts Institute of Technology
Postdoctoral Fellow: Collége de France and University of North Carolina at Chapel Hill
Biography
Matt received his B.A. in Chemistry from Cornell University while working in the laboratory of Pete Wolczanski, where he studied the oxidative addition reactivity of low coordinate Ta(III) complexes with alkyl halides. He continued his education at MIT where, under the guidance of Dan Nocera, he earned his Ph.D. in Inorganic Chemistry working on the synthesis and reactivity of metal oxo complexes within alkoxide platforms. Matt then worked for two years as a post-doctoral research assistant for Marc Fontecave at the Collège de France performing studies that spanned various aspects of CO2 reduction catalyzed by molecular species. He then moved to the UNC to work with Alex Miller. Matt’s work elucidated the mechanisms for the photochemical release of H2 by metal hydride complexes.
Research Summary
The Chambers Group is a synthetic inorganic chemistry laboratory that uses coordination chemistry and ligand design to address central challenges in renewable energy and organometallic catalysis. Projects develop new strategies to promote thermodynamically challenging and kinetically demanding transformation via the synthesis of novel molecular platforms and characterization of reactivity through mechanistic studies. Traditional catalysis focuses on overall reactions with sufficient thermodynamic driving force to proceed. The Chambers Group seeks to integrate catalysts with energy inputs (such as electrical current and light) to mediate reactions that would be otherwise unable to proceed. In this pursuit, a new range of catalytic energy-storing transformations will be developed with impacts in the fields of energy, environmental and organic catalysis.
Students in the Chambers Group will develop expertise in inorganic synthesis, catalysis, photo-chemistry, electrochemistry, crystallography, and an array of spectroscopic techniques. Brief descriptions of the primary research directions are provided below. Students are encouraged to contact Matt for more details regarding these and other projects.
Photoreactive Transition Metal Complexes Containing Metal-Ligand Multiple Bonds
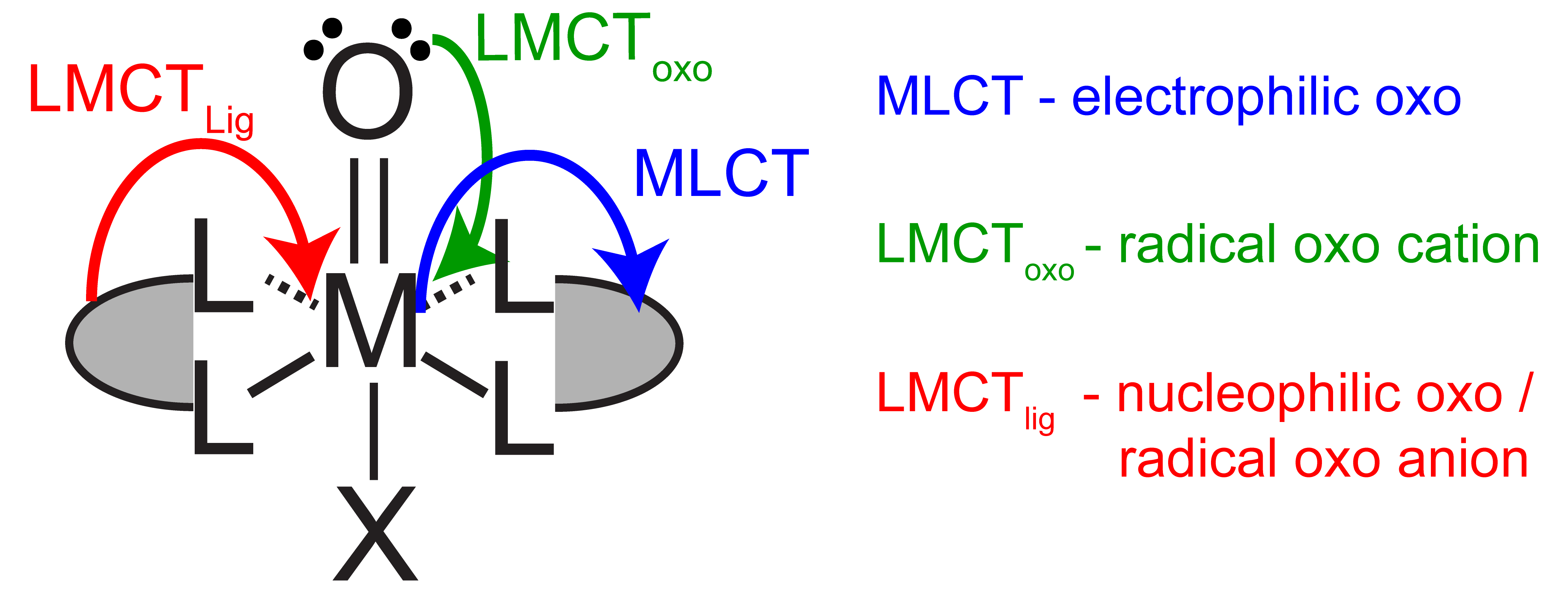
Figure 1. Controlling metal oxo reactivity with excited states
Photochemical reactions have the ability to utilize the thermodynamic driving force and unique electronic structure generated upon photon absorption to directly mediate chemical transformations. However, the development of photocatalytic systems are thwarted by photodegradation pathways due to the propensity of metal-ligand single bonds to homolyze and liberate high energy species that react indiscriminately. Metal-ligand multiple bonds provide motifs that can be exploited in photocatalysis by generating reactive excited states without loss of the multiply-bonded ligand.
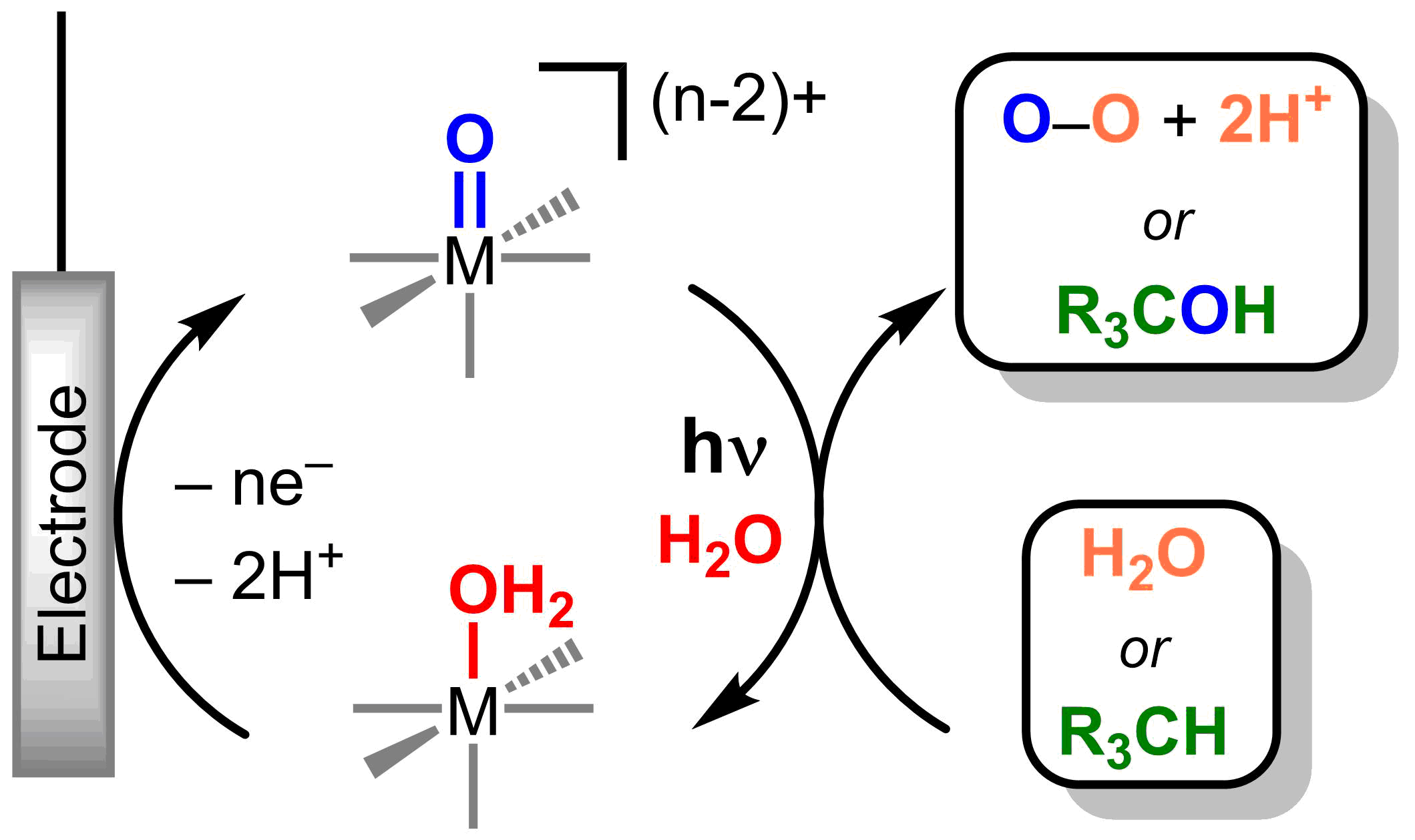
Figure 2. Photoelectrochemical catalytic cycle with metal oxos
The initial focus of the Chambers Group is to develop photocatalysts using metal oxo platforms. Metal oxos have been identified as central intermediates in industrially and biologically relevant transformations. Significant landmarks pertinent to metal oxo reactivity remain, including the activation alkanes, control over selective oxidations for complex substrates, and the efficient oxidation of H2O.
Metal oxos are versatile and can react as electrophiles, nucleophiles, or radicals, resulting in challenges in controlling product selectivity. Reactivity is dictated by the d-electron count and the electronic spin state at the metal. The electronic structure of a metal can be directly manipulated through light absorption and the nature of the excited electronic states can afford tunable reactivity at the metal oxo (Figure 1) without laborious synthetic modifications of ancillary ligand environment. Catalytic systems based on photoreactive metal oxos will be developed through the use of a closed photo-electrochemical cycles (Figure 2).
Reactive Ligand Platforms for Cooperative Bond Activations and Reductions
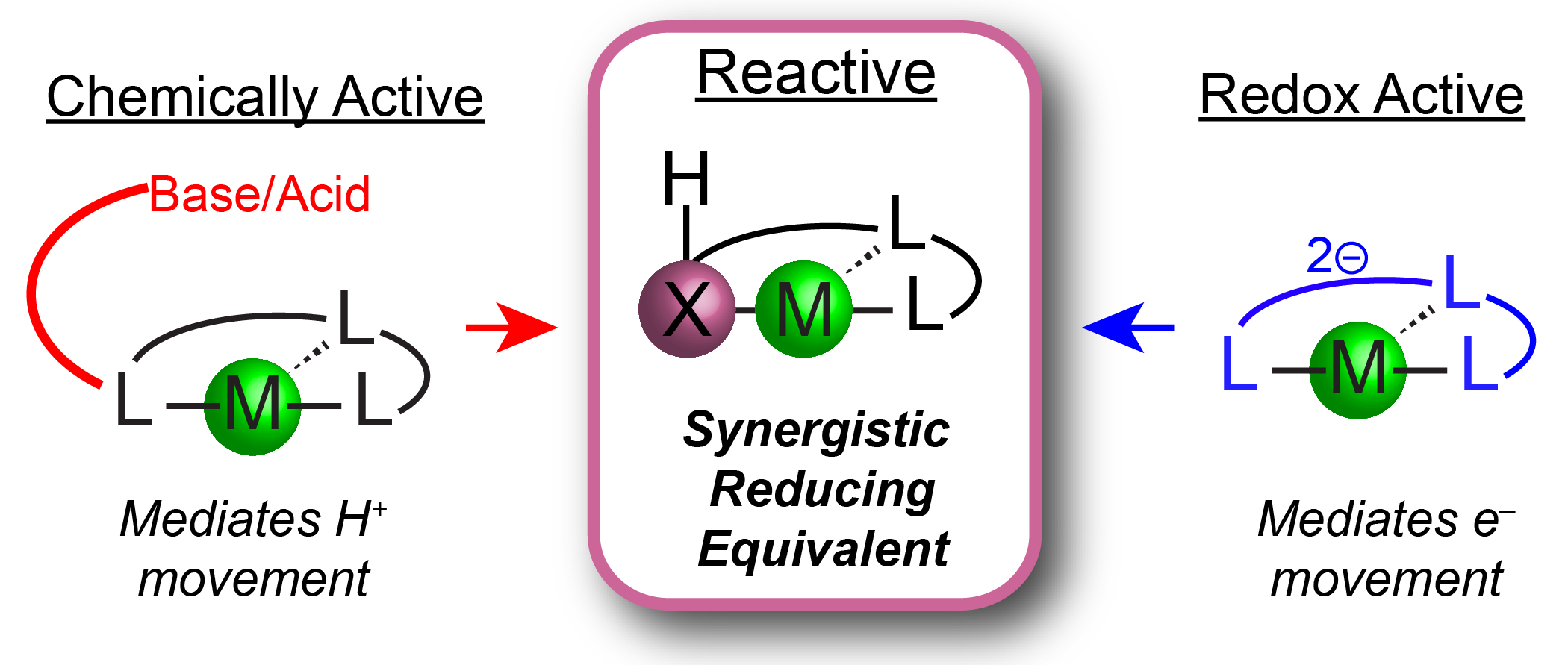 `
`
Figure 3. Mediating H+/e– transfers with reactive ligand scaffolds
Homogeneous molecular catalysts are invaluable in reduction chemistries due to the ability to tune the ancillary ligand field to control reactivity. Prior strategies to modulate reduction reactions include the use of redox active ligands or the presence of chemically active Lewis acidic or basic sites in the secondary coordination sphere. These approaches have independently facilitated reductive processes by storing equivalents of either H+ or e– within the ligand platform which can transfer to the metal center or directly to a metal-bound substrate (Figure 3).
The Chambers Group will combine these strategies and design redox and chemically active ancillary ligands composed of non-first row group 14 and 15 elements, to mediate mult H+/e– reactions and access novel intermediate binding modes. These ligands will contain components with well-defined 2e– redox couples as well as possessing bonds to hydrogen atoms with similar thermodynamic values as organic π bonds. Compared to other types of ligand cooperativity, these ligand scaffolds position the reactive site within the primary coordination sphere of the transition metal complexes to promote synergistic chemistry.
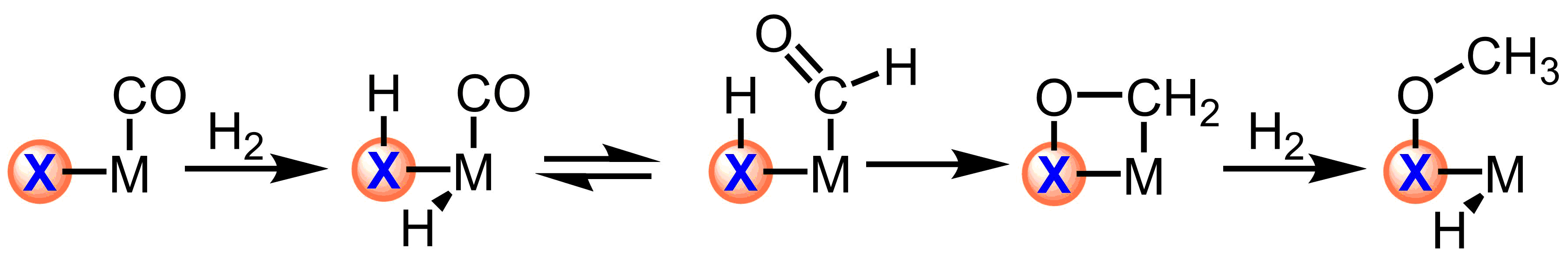
Scheme 1. Trapping migratory insertion products with reactive ligands
The use of reactive ligands based on main group elements will allow for trapping the minor products of equilibria, such as those of migratory insertion. Furthermore, this strategy allows for the independent tuning of the electronic structure of the ligand and metal center with direct applications in controlling the chemoselectivity of outersphere hydrogenations.
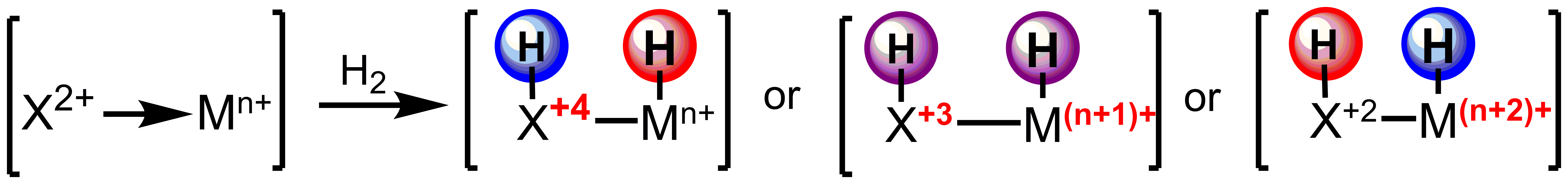
Scheme 2. Tuning acidity and hydricity of reducing equivalents for outersphere hydrogenation
Solution Phase Coordination Chemistry to Control Solid State Surfaces During Catalysis
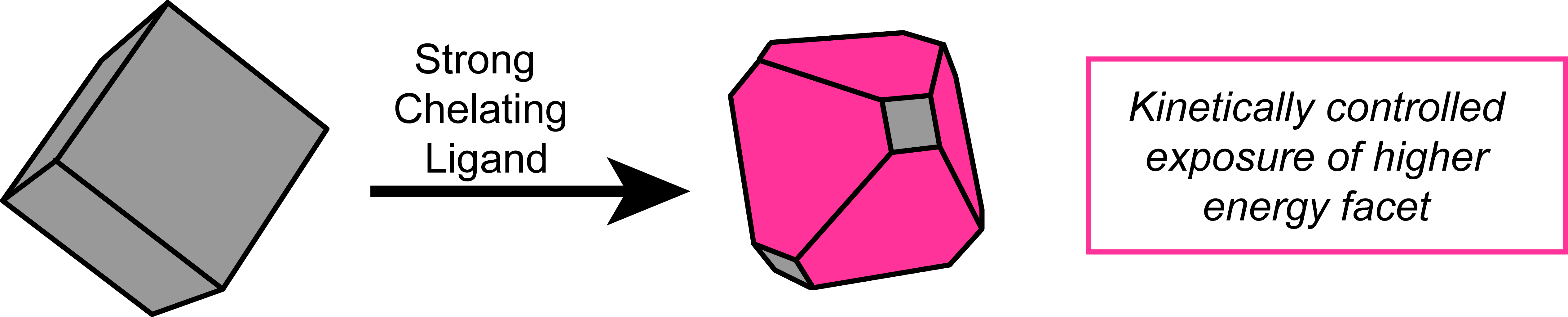
Figure 4. In operando control of catalytic surface sites
The reactivity of solid state catalysts is dictated by the composition, size, and facets of the exposed surface sites. These properties can be finely tuned prior to catalysis. During operation, however, the nature of the surface of solid state catalysts are constantly changing. A key component controlling the manner in which the surfaces evolve is the dissolution equilibria at the solid/liquid interface. The Chambers Group will seek to perturb these underlying dissolution equilibria though the use of rationally designed inert chelating agents in solution. This approach aims to manipulate the active surfaces in operando to afford kinetically accessible metastable phases with the goal of solution phase optimization of solid state catalysts (Figure 4).